Understanding NGS Data Alignment and Best Alignment Tools
1. Introduction
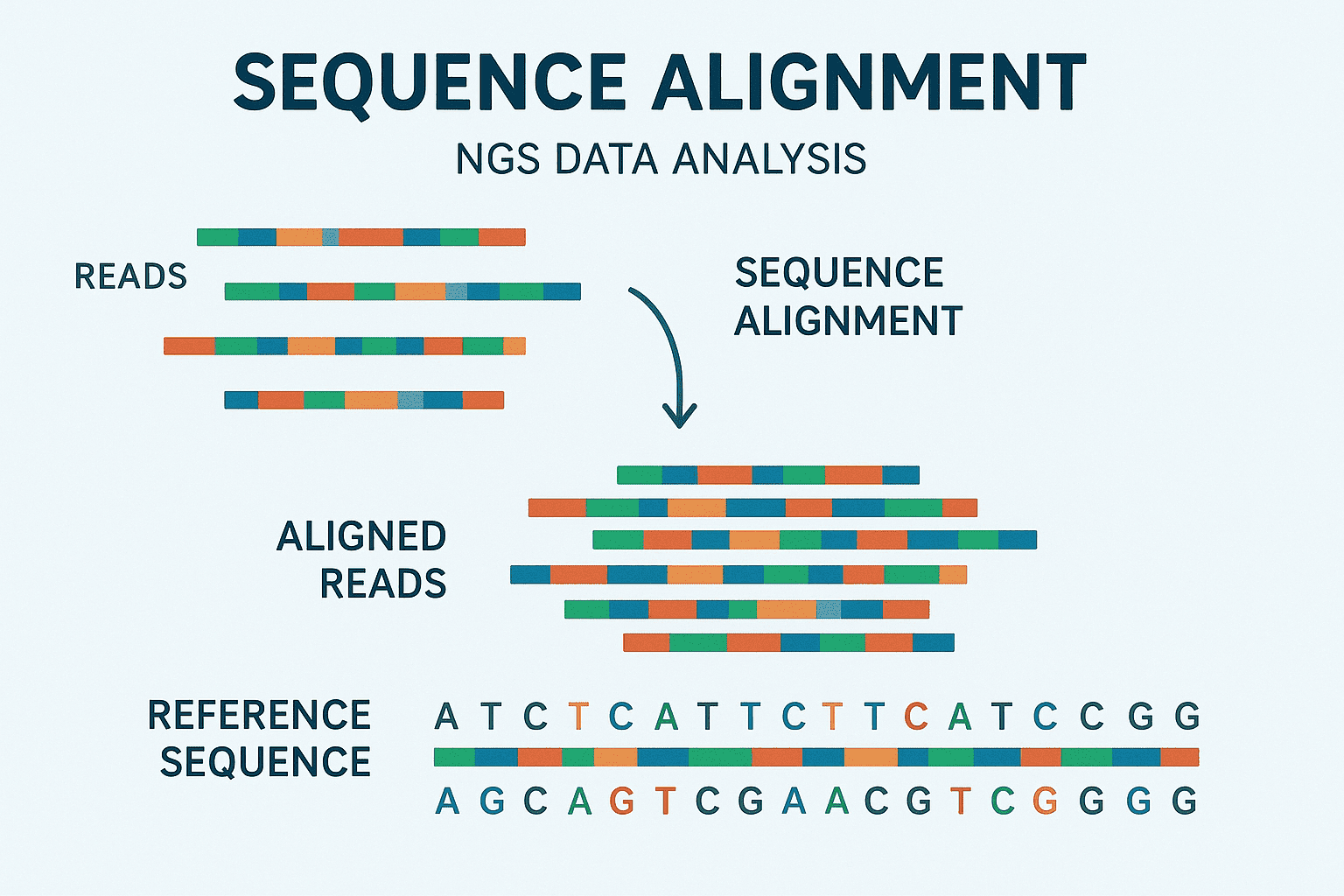
Sequence Alignment plays a vital role in the subsequent analysis of NGS data, where millions of sequenced DNA fragments (reads) need to be aligned with a chosen reference sequence in a timely manner.
The challenge lies in accurately identifying the correct position in the reference genome from which the read originates. Due to the presence of repetitive regions in the genome and the limited length of reads, which range from 50 to 150 bp, it is common for shorter reads to align at multiple locations within the genome.
Additionally, a certain level of flexibility must be permitted for discrepancies with the reference genome during alignment to detect point mutations and other genetic alterations.
Given the vast amount of data produced during NGS analyses, all alignment algorithms utilize supplementary data structures (indices) that facilitate rapid access and matching of sequence data.
These indices can be created either from all generated reads or from the entire reference genome, depending on the algorithm employed. Algorithms from computer science, such as hash tables, or data compression techniques like suffix arrays, are frequently utilized in alignment tools.
With these algorithms, it is feasible to compare over 100 GB of sequence data from NGS analyses against the human reference genome within just a few hours. Furthermore, by leveraging extensive parallel computing power (CPUs), this processing time can be significantly reduced.
Consequently, even substantial volumes of sequencing data from whole-exome or whole-genome sequencing can be processed efficiently.
2. Alignment Definition
The subsequent phase involves aligning your sequencing reads to a reference genome or transcriptome. A significant challenge to consider is the sheer size and complexity of the human genome.
Sequencers can generate billions of reads in a single run, and they are susceptible to errors. Therefore, achieving precise alignment is a labor-intensive endeavor.
Sequence databases such as GenBank (http://www.ncbi.nlm.nih.gov/genbank/) experienced rapid growth during the 1980s, making a comprehensive dynamic programming comparison of any query sequence against every known sequence computationally expensive. As a result, the need to align a query sequence with a database led to the creation of a heuristic algorithm, which was incorporated into the FASTA program suite.
The fundamental concept of this algorithm is to eliminate large sections of the database from the costly dynamic programming comparison by swiftly identifying candidate sequences that have short segments (k-tuples) of highly similar sequence with the query.
Following FASTA, the BLAST program emerged, offering additional speed benefits and a new feature that estimates the statistical probability that each matching sequence was found by chance. BLAST remains one of the most prevalent search tools for biological sequence databases.
With the advent of ultra-high-throughput sequencing technologies in 2007, new alignment challenges surfaced. This chapter outlines these advancements and the current leading practices in NGS alignment algorithms. Computational biologists have developed over 70 read mapping methods to date.
Sequence alignment is extensively utilized in molecular biology to identify similar DNA, RNA, or protein sequences. These algorithms typically fall into two categories: global (Needleman–Wunsch), which aligns the entire sequence, and local (Smith–Waterman), which focuses solely on finding highly similar subsequences.
3. Global Alignment (Needleman–Wunsch Algorithm)
Statistically, the potential solutions are vast; nevertheless, our focus is on achieving optimal alignments with minimal discrepancies such as indels or mismatches. The term unit edit distance (edist) refers to the count of mismatches, insertions, and deletions present in an optimal sequence alignment.
The primary objective is to reduce the edist by organizing partial solutions within a (m+1) x (n+1) matrix. Assuming that both input sequences a and b originate from the same source, a global alignment seeks to locate corresponding segments and the modifications required to convert one sequence into the other.
These modifications are assigned scores, and an optimal set of changes is determined, which outlines an alignment. The dynamic programming method organizes optimal subsolutions in matrix E, where an entry E(i,j) signifies the best score for aligning the prefixes a1..i with b1..j.
The scoring matrix created using the Needleman–Wunsch algorithm, along with the associated traceback matrix, facilitates the discovery of the best alignment. An example of one possible alignment outcome along with its corresponding traceback is provided.
4. Local Alignment (Smith–Waterman Algorithm)
Local alignment conducted by the Smith–Waterman algorithm seeks to identify similarities between two sequences of nucleic acids or proteins. The primary distinction from global alignment is that cells in the scoring matrix that yield negative values are set to zero. To better grasp local or sub-region alignment, consider a small Toy genome (16 bp): CATGGTCATTGGTTCC.
Local alignment utilizes a hash-based method with two primary strategies: hashing the reference and employing the Burrows–Wheeler transform. The initial step involves hashing or indexing the genome (only the forward strand), which results in a hash/k-mer index of your Toy genome.
Now, you aim to align a Toy sequencing read (TGGTCA) to this indexed Toy genome. The k-mer index can be utilized to swiftly locate candidate alignment positions in the reference genome. For instance, the k-mer TGG corresponds to positions 3 and 10, while the k-mer TCA relates to position 6.
Consequently, the Burrows–Wheeler transform serves as an alternative method for performing precise matches on hashes, comparing them against the genome and calculating a score.
This method endeavors to pinpoint the most similar subsequences that optimize the scoring of their corresponding segments and the modifications required to convert one subsequence into another. The dynamic programming technique organizes optimal subsolutions within matrix E, where an entry Ei,j denotes the maximum similarity score for any local alignment of the (sub)prefixes ax..i with by..j, where x and y are both greater than zero and must be determined through traceback.
Note: consecutive gap (Indels) scoring is conducted linearly.
Alignment to a reference genome can be carried out with either single-end or paired-end sequencing reads, depending on the specifics of your experiment and library preparation. Paired-end sequencing is advisable for RNA-Seq experiments.
Additionally, we distinguish between two categories of aligners:
• Splice unaware
• Splice aware
Splice-unaware aligners can align continuous reads to a reference genome, but they do not recognize exon/intron junctions. Therefore, in RNA sequencing, splice-unaware aligners are not suitable for analyzing the expression of known genes or aligning reads to the transcriptome.
Conversely, splice-aware aligners map reads across exon/intron junctions and are thus utilized for identifying new splice variants, as well as for analyzing gene expression levels.
In this context, the most frequently used alignment tools will be discussed in the following section.
5. Alignment Tools
5.1 STAR
Spliced Transcripts Alignment to a Reference (STAR) is a standalone application that employs a sequential maximum mappable seed search followed by seed clustering and stitching techniques to align RNA-Seq reads. It can identify canonical junctions, non-canonical splicing events, and chimeric transcripts.
The primary benefits of STAR include its rapid processing, precision, and effectiveness. It is developed as standalone C++ software and is available for free on GitHub (https://github.com/alexdobin/STAR/releases).
5.2 Bowtie
Bowtie is an extremely fast and memory-efficient tool for aligning short reads to large reference genomes that have been indexed using a Burrows-Wheeler index. It is commonly utilized for aligning DNA sequences, as it does not account for splicing. This characteristic makes it particularly useful in microbiome alignment (http://bowtie-bio.sourceforge.net/index.shtml).
5.3 Bowtie2
Bowtie2, like Bowtie, is also an ultrafast and memory-efficient tool, but it is better suited for aligning sequencing reads ranging from 50 to thousands of characters to longer reference sequences (e.g., mammalian genomes) indexed with a Ferragina–Manzini (Fm) index. Bowtie2 offers support for gapped, local, and paired-end alignment modes (https://github.com/BenLangmead/bowtie2).
5.4 TopHat/TopHat2
TopHat aligns RNA-Seq reads to genomes by initially utilizing the short-read aligner Bowtie, and subsequently mapping to a reference genome to identify RNA splice sites de novo. RNA-Seq reads are aligned against the entire reference genome, with unmapped reads being discarded. TopHat is frequently used in conjunction with the Cufflinks software for comprehensive sequencing data analysis (https://github.com/dnanexus/tophat_cufflinks_RNA-Seq/tree/master/tophat2).
A thorough explanation of how to use TopHat can be found in the TopHat manual (http://ccb.jhu.edu/software/tophat/manual.shtml).
5.5 Burrow–Wheeler Aligner (BWA)
BWA is a splice-unaware software suite designed for mapping low-divergent sequences against large reference genomes, such as the human genome. It comprises three algorithms: BWA-backtrack, BWA-SW, and BWA-MEM.
The first algorithm is tailored for Illumina sequence reads of up to 100bp. BWA-MEM and BWA-SW both provide features like long-read support and split alignment, but BWA-MEM (maximal exact matches), the most recent development, is typically favored for high-quality queries due to its speed and accuracy (https://github.com/lh3/bwa).
The splice-unaware alignment algorithms are suggested for organisms like bacteria. A comprehensive guide on using BWA can be found in the BWA manual (http://bio-bwa.sourceforge.net/bwa.shtml).
5.6 HISAT2
HISAT (along with its upgraded version HISAT2) represents the next evolution of spliced aligners developed by the same team behind TopHat.
HISAT employs an indexing method derived from the Burrows–Wheeler transform and the Ferragina–Manzini (Fm) index, utilizing two types of indices for alignment: a comprehensive genome Fm index to anchor each alignment and multiple local Fm indices for extremely fast extensions of these alignments (https://github.com/DaehwanKimLab/hisat).
Some of the most notable features of HISAT include its impressive speed and minimal memory usage. HISAT is open-source software that is available at no cost. For a comprehensive guide on how to use HISAT, refer to the HISAT manual (https://ccb.jhu.edu/software/hisat2/manual.shtml).
6. Conclusion
• Sequence alignment refers to the method of assessing and identifying distances/similarities among biological sequences.
• The dynamic programming approach can be utilized for global alignments through techniques like global and local alignment algorithms.
• The metric that quantifies the level of sequence similarity is known as the alignment score.
• Sequence alignment involves determining the so-called edit distance, which typically reflects the smallest number of substitutions, insertions, and deletions required to transform one sequence into another.
• The selection of a sequencing read alignment tool is influenced by your objectives and the particular circumstances.
7. FAQs
1) What is alignment in NGS?
Alignment is the process of arranging DNA/RNA/protein sequences so that homologous characters line up. In NGS, it most often means placing short or long reads onto a reference genome (read mapping) or aligning two assemblies (genome-to-genome) so we can call variants, quantify expression, or compare genomes.
2) What are the main types of sequence alignment?
Global (Needleman–Wunsch): end-to-end; best when sequences are similar and full-length.
Local (Smith–Waterman): finds the best matching subsections (motifs, domains).
Semi-global/Overlap: ignores terminal gaps (great for adapters/overlaps).
Spliced alignment: allows introns; used for RNA-seq.
Multiple sequence alignment (MSA): aligns 3+ sequences to study conservation/phylogeny.
3) What’s the difference between mapping and alignment?
“Mapping” answers where a read best fits on a reference using a fast index (BWT/FM-index or minimizers). The mapper then emits an alignment: a base-by-base description (CIGAR string) of matches, mismatches, gaps, and—for RNA-seq—splices. In practice, mappers produce alignments; the terms are often used interchangeably.
Recent Posts
Sequence Alignment plays a vital role in the subsequent analysis of NGS data, where millions of sequenced DNA fragments (reads) need to be aligned with a chosen reference sequence in a timely manner.
FASTQ files serve as the “raw data files” for any sequencing application, indicating that they are “unaltered.” Consequently, this file format is utilized for performing Quality Checks on sequencing reads. The Quality Check process is typically carried out using the FastQC tool developed by Simon Andrews from Babraham Bioinformatics.
FASTA format is a text-oriented format utilized for depicting either nucleotide sequences or peptide sequences, where nucleotides or amino acids are denoted by a single-letter code.
Mammalian expression systems enable the production of complex, functional recombinant proteins with proper folding and post-translational modifications. These systems are ideal for studying human proteins in a near-native environment, offering advantages in scalability, gene delivery, and purification. HEK293 and CHO cells remain the most widely used hosts, supporting both transient and stable expression strategies for academic and pharmaceutical applications.
Gas Chromatography (GC) stands as one of the most powerful and versatile analytical techniques used to separate and analyze compounds in complex mixtures. At its core, GC enables the identification and quantification of chemical substances based on their molecular composition and retention behaviors during migration through a chromatographic column.